
Introduction
Neurons, the fundamental units of the nervous system, are highly polarized cells distinguished by their unique morphology: a single, long axon and multiple branching dendrites. This polarization is crucial for their function in receiving, processing, and transmitting information. Underpinning this intricate cellular architecture is the microtubule cytoskeleton, a dynamic network of polymers essential for maintaining cellular integrity, intracellular transport, and overall neuronal function (Conde and Cáceres, 2009; Kapitein and Hoogenraad, 2015). Microtubules themselves possess an intrinsic polarity, with a plus end and a minus end, which dictates the directionality of motor protein movement. These motor proteins, such as kinesins and dyneins, recognize this asymmetry, enabling the targeted transport of diverse cellular cargoes along the microtubule network (Hirokawa et al., 2010; Maday et al., 2014). The precise organization of microtubules within axons and dendrites is therefore paramount for directing polarized cargo trafficking and establishing neuronal identity (Kapitein and Hoogenraad, 2011; Rolls, 2011).
Research in hippocampal neurons has illuminated the role of microtubule polarity in axonal transport. Kinesin-1, a plus-end-directed motor, selectively transports cargo into the axon along microtubules oriented with their plus ends distal to the cell body, often referred to as “plus-end-out” orientation (Nakata and Hirokawa, 2003; Hammond et al., 2010; Kapitein et al., 2010a; Nakata et al., 2011; Huang and Banker, 2012). Conversely, the selective presence of “minus-end-out” oriented microtubules in dendrites allows dynein, a minus-end-directed motor, to selectively deliver cargo to these compartments (Kapitein et al., 2010b). While the significance of microtubule organization for intracellular trafficking is well-established, the detailed arrangement of microtubules in axons and dendrites, particularly during neuronal development and in mature neurons within complex tissue environments, remains a subject of ongoing investigation.
Early investigations using the hook-decoration technique and electron microscopy (EM) laid the groundwork for our understanding of microtubule polarity in neurons (Baas and Lin, 2011). These studies revealed a striking contrast: axons of cultured vertebrate neurons exhibit a uniform microtubule arrangement, with virtually all microtubules oriented plus-end-out (Baas et al., 1987; Baas et al., 1988). In stark opposition to this uniformity, proximal dendrites were found to contain a non-uniform, mixed population of microtubules, approximately half plus-end-out and half minus-end-out (Baas et al., 1988; Burton, 1988). While some studies suggested a shift towards a predominantly plus-end-out orientation in distal dendrites of hippocampal neurons, the prevailing view highlighted the heterogeneous microtubule organization as a defining feature of dendrites, distinct from the homogenous axonal microtubule array (Baas et al., 1988; Burton, 1988). Fluorescently labeled microtubule plus-end binding proteins in live hippocampal and Purkinje cell cultures further corroborated the existence of mixed microtubule polarities in dendrites, although the proportion of minus-end-out microtubules appeared to be lower than initially estimated by EM (Stepanova et al., 2003). Furthermore, evidence for diverse microtubule arrays in mouse brain tissue has emerged from second-harmonic generation microscopy and live-cell imaging of microtubule growth (Kwan et al., 2008; Kleele et al., 2014). Intriguingly, invertebrate neurons, such as those in Drosophila and Caenorhabditis elegans, present a different picture, with dendrites primarily characterized by minus-end-out microtubules, although mixed orientations have also been observed (Stone et al., 2008; Goodwin et al., 2012; Maniar et al., 2012). These findings underscore the notion that the presence of minus-end-out oriented microtubules, and thus a lack of uniformity in microtubule polarity, may be a distinctive characteristic of dendrites.
This study delves deeper into the organization of microtubules in axons and dendrites, employing a multifaceted approach combining cell biology, quantitative microscopy, laser microsurgery, organotypic slice cultures, and in vivo imaging in living mice. Our aim was to rigorously determine microtubule orientations and dynamics in diverse neuronal contexts. Our findings robustly demonstrate that mature dendrites, in vitro, ex vivo, and in vivo, are characterized by non-uniformly oriented microtubules. Crucially, we reveal an approximately equal distribution of plus-end-out and minus-end-out microtubule orientations throughout mature dendritic processes. Intriguingly, our investigations into early neuronal development uncover the presence of minus-end-out microtubules in nascent neurites even before neuronal polarization occurs. These data suggest that the mixed, non-uniform microtubule polarity is established early in neuronal development, preceding axon formation and neuronal polarization. Based on these comprehensive observations, we propose a model where the selective formation of uniform plus-end-out microtubules in the axon is a critical and active process that underpins neuronal polarization, highlighting the functional significance of the contrast between axonal uniformity and dendritic non-uniformity in microtubule organization.
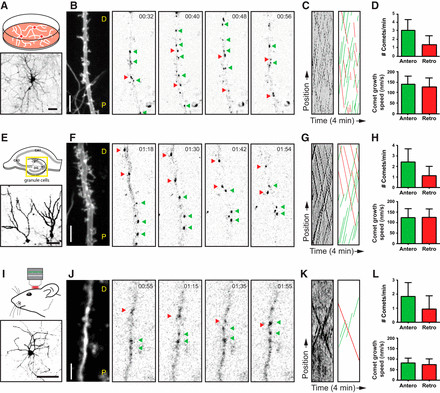 Figure 1.
Figure 1.
Materials and Methods
Animals
All animal experiments adhered to the guidelines for the welfare of experimental animals issued by the government of The Netherlands or the Swiss Federal Act on Animal Protection. Protocols were reviewed and approved by the Animal Ethical Review Committee of Utrecht University or the University of Geneva and the Geneva Cantonal Veterinary Office.
DNA constructs
The CAMSAP2–shRNA (ttgcatgtgctcaacagt) construct was previously described (Yau et al., 2014). The GFP–MT+TIP reporter, designed to visualize microtubule plus ends, incorporates the SxIP motif from human MACF2, recognized by end-binding (EB) proteins. It was created by fusing a two-stranded leucine zipper coiled-coil sequence corresponding to GCN4-p1 (RMKQLEDKVEELLSKNYHLENEVARLKKLVGER) to the N-terminal 18 amino acids of human MACF2 (Gly5468-Lys5485; NP_899236) using PCR-based techniques (Honnappa et al., 2009). A glycine-rich linker (GAGG) was inserted between GCN4 p1 and MACF18 and subcloned into the pβactin-16 pl expression vector to generate pβactin–GFP–GCN4–MACF18. For experiments in stage 2–3 neurons, MACF43 was used, identical to MACF18 but including the 43 N-terminal amino acids of human MACF2 (Glu5455–Arg5497). This GFP–GCN4–MACF18/43 construct served as a general marker for microtubule growing plus ends and was termed GFP–MT+TIP. The pβactin–GFP and pβactin–mRFP plasmids have been described previously (Kapitein et al., 2010a). The volume marker MARCKS–TagRFP-T was adapted from MARCKS–GFP (De Paola et al., 2003. The first 41 amino acids of MARCKS (MGCQFSKTAAKGEAAAERPGEAAVASSPSKANGQENGHVKV), containing an Ala-Cys mutation, were fused to TagRFP-T via a GS-linker (PCR). MARCKS–TagRFP-T was cloned upstream and GFP–GCN4–MACF18 downstream of an attenuated ECMV internal ribosome entry site (pIRES) in a bicistronic construct, resulting in high expression of the volume marker and low expression of GFP–MT+TIP. This bicistronic construct was subcloned into either the pCAGGS vector for single-cell electroporation or the lentivirus vector pSIN–TRE–mSEAP–hPGK–rtTA2sM2 (provided by Dr. Didier Trono, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland). The human PGK promoter was replaced with a short rat synapsin (0.5 kb) promoter to restrict expression primarily to neurons. Lentiviral particles were produced as previously described (Yau et al., 2014).
Primary cortical and hippocampal neuron cultures, transfection, and infections
Primary cortical and hippocampal cultures were derived from embryonic day 18 rat brains (of either sex). Cells were plated at a density of 75,000/well on coverslips coated with poly-L-lysine (37.5 μg/ml) and laminin (1.25 μg/ml). Dissociated neurons were cultured in Neurobasal medium (NB) supplemented with 2% B27 (Gibco), 0.5 mM glutamine (Gibco), 15.6 μM glutamate (Sigma), and 1% penicillin/streptomycin (Gibco).
Transfections
Hippocampal neurons were transfected using Lipofectamine 2000 (Invitrogen). Briefly, DNA (1.8 μg/well, for a 12-well plate) was mixed with 3.3 μl of Lipofectamine 2000 in 200 μl of NB, incubated for 30 min, and added to neurons in NB at 37°C in 5% CO2 for 45 min. Neurons were then washed with NB and returned to their original medium at 37°C in 5% CO2 for 2–4 days.
Nucleofections
Primary cortical neurons (1.3 × 10^6 cells) were nucleofected with 0.5 μg of GFP–MACF43 or GFP plasmid and 2.5 μg of empty plasmid or shRNA using the Amaxa Rat Neuron Nucleofector kit (Lonza) following manufacturer’s instructions. Cells were cultured for 18–24 h at 37°C in 5% CO2 before imaging.
Lentivirus infections
Hippocampal neurons were transduced with lentivirus 7–10 days prior to experiments. Tetracycline-dependent expression was induced by supplementing the medium with 500 ng/ml doxycycline 2 days before imaging.
Organotypic slice cultures and lentivirus infections
Mouse hippocampal slices (350 μm) were prepared from postnatal day 5–6 pups (of either sex). Slices were transferred onto small patches of FHLC membrane (0.45 μm; Millipore) positioned on Millicell culture inserts (Millipore). Hippocampal slices were transduced with lentivirus immediately after plating by applying a drop of concentrated virus solution over the culture. Medium exchange and maintenance of cultures were performed as previously described (Stoppini et al., 1991). Experiments were conducted after 14–21 days in vitro (DIV). Slice culture medium was supplemented with doxycycline (500 ng/ml) at least 5 days before imaging.
In vivo single-cell electroporation
In vivo single-cell DNA electroporation was performed as described previously (Pagès et al., 2015). Briefly, young male mice (4–6 weeks old) were anesthetized with an intraperitoneal injection of MMF [a mixture of medetomidin (Dorbene, 0.2 mg/kg), midazolam (Dormicum, 5 mg/kg), and fentanyl (Duragesic, 0.05 mg/kg) in saline]. A craniotomy was performed above the somatosensory cortex. A 15–20 MΩ glass pipette (GC150F-7.5; Harvard Apparatus) was filled with internal solution [in mM: 266 KMeSO4, 14 KCl, 20 Na-HEPES, 4 MgATP, 4 Na2ATP, 1 Na2GFP, and 0.1 EGTA, pH 7.2 (280–290 mOsm)], containing 30 ng/μl plasmid DNA and Alexa Fluor 488 hydrazide (50 μM; Life Technologies). Under visual guidance, cortical layer 2/3 (L2/3) pyramidal cells were targeted and electroporated (10 pulses, −12 V, 500 μs, 50 Hz) using a head stage (AP-1AX1MU) attached to an Axoporator 800A (Molecular Devices). Successful electroporations resulted in rapid filling of cell bodies with Alexa Fluor 488 hydrazide. The pipette was then gently withdrawn, and a glass coverslip (3 μm diameter) was implanted to cover the craniotomy. Imaging commenced after 1 week of recovery.
Live-cell confocal imaging and laser-induced severing
Spinning-disk confocal microscopy was conducted on an inverted microscope (Nikon Eclipse Ti with Perfect Focus System) equipped with a Plan Apo VC 100×, 1.4 numerical aperture (NA) oil-immersion objective or an S Fluor 100×, 0.5–1.3 NA oil-immersion objective (Nikon) for laser-induced severing (LS) experiments. MetaMorph software controlled the Evolve 512 EMCCD camera (Photometrics) and all motorized components. The microscope was further equipped with an ASI motorized stage MS-2000-XYZ with piezo top plate, ILas system (Roper Scientific France/PICT-IBiSA, Curie Institute), and Shutter LB10-3. Fluorescence excitation was achieved using a Calypso 491 nm, 100 mW laser and Jive 561, 100 mW laser (Cobolt). A Teem Photonics 355 nm Q-switched pulsed laser was used for LS (Botvinick et al., 2004; Colombelli et al., 2005). Dichroic filters ET-GFP/mCherry (59022; Chroma Technology) or sequential filters ET-GFP (49002; Chroma Technology) and ET-mCherry (49008; Chroma Technology) were used for wavelength selection. All imaging was performed in full conditioned medium for hippocampal neuron cultures. A Tokai Hit Stage Top Incubator (INUBG2E-ZILCS) maintained neurons at 37°C with 5% CO2.
Imaging stage 1–2 neurons
Time-lapse acquisition was performed for 6 min (without LS) or for 1 min before LS and 5 min after, with a time interval of 1 s. LS was performed at 10 μm from the soma. Regions of 10 μm before and after the LS position were used for quantifications. In neurites shorter than 20 μm, LS was performed at the neurite midpoint.
Imaging stage 3–5 neurons
Microtubule plus-tip imaging in neuron cultures was performed with 6 × 0.5 μm steps and sequential channel recordings. Time-lapse recordings were performed in a single plane when LS was conducted sequentially.
Imaging taxol-treated neurons
Hippocampal neurons were incubated with DMSO or 10 nM taxol at DIV1 for 72 h. For microtubule LS experiments, control and taxol-treated neurons were transfected at DIV3 with GFP–MT+TIP using Lipofectamine 2000 (Invitrogen). At DIV4, neurons were fixed for immunostaining or imaged for live-cell experiments.
Imaging organotypic slices
Slice cultures were imaged in ACSF [126 mM NaCl, 3 mM KCl, 2.5 mM CaCl2, 1.3 mM MgCl2, 1.25 mM Na2HPO4, 26 mM NaHCO3, 20 mM glucose, and 1 mM Trolox (bubbled with 95% O2, 5% CO2)]. Dendrites were imaged with z-stacks of 6–8 × 0.5 μm steps, except for LS experiments (single plane). Imaging positions along dendrites were defined as proximal (within 60% of total dendritic length from the cell body).
In vivo two-photon laser scanning microscopy
Imaging was performed using a custom-built two-photon laser-scanning microscope (https://openwiki.janelia.org/wiki/display/shareddesigns/Shared+Two-photon+Microscope+Designs) and Scanimage data acquisition software (https://openwiki.janelia.org/wiki/display/ephus/ScanImage). Anesthetized mice (with MMF; see above) were head-fixed under the microscope, and body temperature was maintained using a feedback-controlled heating pad. Fluorophores were excited using a titanium:sapphire laser (Chameleon Ultra II; Coherent) tuned at 950 nm. Emitted fluorescence was collected using a 16× multiphoton objective (0.8 NA, N16XLWD-PF; Nikon), spectrally separated using two bandpass filters (HC530/55, HC607/70, BrightLine; Semrock), and detected using two photomultiplier tubes (10770PB-40 for green, and R3896 for red; Hamamatsu). Images were recorded at 0.2 Hz (512 × 512 pixels, 2 ms/line, 5 s interimage interval). Typical fields of view ranged from 30–50 μm square. The average excitation power was kept below 50 mW as measured under the objective.
Immunofluorescence staining
Neurons were fixed in ice-cold 100% methanol/1 mM EGTA for 10 min at −20°C. Following fixation, cells were washed three times for 5 min in PBS and incubated with primary antibody in GDB buffer (0.2% BSA, 0.8 M NaCl, 0.5% Triton X-100, and 30 mM phosphate buffer, pH 7.4) overnight at 4°C. After extensive washing with PBS, neurons were incubated with secondary antibodies in GDB buffer for 1 h at room temperature and washed again. Coverslips were mounted in Vectashield (Vector Laboratories) and sealed with nail polish. The following primary and secondary antibodies were used: rabbit anti-CAMSAP2 (17880-1-AP; Proteintech), mouse anti-α-tubulin (B-5-1-2; Sigma), mouse anti-Tau (MAB3420; Millipore Bioscience Research Reagents), βIII tubulin (PRB-435P; Covance), mouse anti-GFP (1814460; Roche), and Alexa Fluor 488- and Alexa Fluor 568-conjugated secondary antibodies (Invitrogen).
Image processing and quantifications
Image analysis was performed using FIJI, and data processing and statistical analysis were conducted in Excel and GraphPad Prism (GraphPad Software).
MT+TIP analysis
Time-lapse recordings were low-pass filtered and subtracted with an average projection of the complete time lapse (Kapitein et al., 2010b). Kymography was performed on filtered images using the FIJI “KymoResliceWide” plugin. Starting and end positions of traces were marked using the “Cell Counter” plugin, and coordinates were exported for further analysis. The number of microtubule plus-tip comets, comet growth speed, and distances to the cutting position were calculated from the starting and end coordinates of microtubule traces. To ensure comparable data of microtubule plus-tip dynamics across different model systems, comets detectable over 1 min within 20 μm of axonal or dendritic segment were quantified (unless otherwise indicated). In LS experiments, the cut site was positioned in the center of this observation area.
MT stability analysis
Imaging and analysis were performed as described for MT+TIP analysis. The starting points of comets relative to the LS site within a 1-minute time window (before or after LS) were binned into 2 μm intervals and averaged for each DIV15 dendrite or axon. Multiple minutes were averaged for each dendrite and axon after LS. The median value indicates 50% of the data points.
Analysis of CAMSAP2 distribution
Images from a wide-field fluorescence microscope (Eclipse 80i; Nikon) were used to quantify CAMSAP2 distribution. Localization of CAMSAP2 was classified as “tip” (only at the neurite tip), “equal” (homogenous distribution along the neurite), or “random” (scattered accumulations along the neurite).
Analysis of CAMSAP2 staining
Cortical neurons were nucleofected with GFP and either pSuper control or CAMSAP2 shRNA before plating. Neurons were fixed 72 h after transfection and stained for CAMSAP2. Confocal images were acquired using a Zeiss LSM 700. Mean intensities of CAMSAP2 signals were quantified in neurites of control and CAMSAP2-depleted neurons at a distance of 15 μm from the soma using FIJI software.
Results
Mixed Microtubule Organization in Dendrites: A Consistent Feature In Vitro and In Vivo
To investigate the microtubule organization within vertebrate dendrites across different experimental systems, we conducted a systematic analysis of microtubule plus-end dynamics and orientations in primary cultures of rat hippocampal neurons, mouse organotypic hippocampal slice cultures, and layer 2/3 pyramidal neurons in the somatosensory cortex of living mice. We began by performing two-color live-cell imaging in differentiated (DIV18) hippocampal neurons in culture. We utilized a bicistronic lentiviral construct to express both the microtubule plus-end marker GFP–MT+TIP (Yau et al., 2014) and MARCKS–TagRFP, which served to delineate neuronal morphology. In neurons exhibiting low expression levels, we observed a clear distinction in microtubule movement patterns: axons displayed unidirectional displacement of GFP–MT+TIP comets, whereas dendrites exhibited bidirectional movements, reflecting the fundamental difference in microtubule organization between these compartments (Stepanova et al., 2003; Jaworski et al., 2009).
Consistent with prior studies using EB3–GFP as a +TIP marker (Jaworski et al., 2009; Kapitein et al., 2011), we measured a total comet number of 4.4 ± 1.6 comets/min/20 μm (mean ± SD; n = 32 dendrites) and an average comet velocity of 132 ± 53 nm/s (mean ± SD; n > 600 comets) in DIV18 cultured hippocampal neurons. We then focused on the directionality of GFP–MT+TIP comets within middle segments of primary dendrites, defined as the half-length of the dendrite measured from the cell body to the dendritic tip (Fig. 1A–C; Movie 1). Notably, we found that, on average, 31% of labeled plus ends exhibited retrograde movement, directed towards the cell body, while the comet growth speed remained statistically similar in both directions (Fig. 1D). These results definitively indicate that approximately one-third of the dynamic microtubules in dendrites of cultured hippocampal neurons are oriented with their plus ends pointing inward, highlighting the non-uniform microtubule organization in these neuronal processes.
Figure 1.
Expanding our investigation, we examined microtubule dynamics and growth direction in mouse organotypic hippocampal slice cultures. Using the same live-cell imaging approach, we focused on the dendrites of granule cells, the primary neuronal population of the dentate gyrus (Fig. 1E–G; Movie 1). Analogous to our findings in dissociated hippocampal neurons, middle segments of granule cell dendrites displayed, on average, 32% of GFP–MT+TIP comets moving retrogradely towards the cell body (Fig. 1H). In slice cultures, we observed a slight reduction in both the total number of comets and comet growth speed in both directions compared to dissociated cultures (Fig. 1H), potentially reflecting the more complex and constrained environment within tissue slices.
To further validate our findings in a more physiologically relevant context, we extended our analysis to in vivo conditions, recording microtubule dynamics and orientation in dendrites of layer 2/3 pyramidal cells in the somatosensory cortex of living mice (Fig. 1I–K; Movie 1). The bicistronic GFP–MT+TIP and MARCKS–TagRFP construct was introduced into the neocortex of 6-week-old mice via single-cell electroporation, and in vivo two-photon laser scanning microscopy (2PLSM) imaging was performed through a cranial window (Cane et al., 2014; Pagès et al., 2015). Neurons exhibiting sufficient brightness in both channels were selected for high-magnification imaging. Consistent with our in vitro and ex vivo observations, we observed clear bidirectional displacements of GFP–MT+TIP comets in dendrites in vivo. Similar to the culture systems, pyramidal cell dendrites in vivo exhibited an average of 34% of GFP–MT+TIP comets moving retrogradely towards the cell body (Fig. 1L). Notably, the total number of comets and comet growth speed were further reduced in dendrites in vivo compared to in vitro systems (Fig. 1L), likely reflecting the highly complex and densely packed environment of the living brain.
Collectively, these data provide compelling evidence that a mixed, non-uniform microtubule organization is a consistent feature of mature dendrites across different neuron types and experimental systems, encompassing in vitro, ex vivo, and in vivo conditions. While the dynamics of microtubules, as indicated by comet number and growth speed, are modulated by the cellular environment, the fundamental ratio of dynamic plus-end-in (approximately 1:3) and plus-end-out (approximately 2:3) microtubules in dendrites appears to be a remarkably conserved parameter across all analyzed model systems, reinforcing the notion that dendritic microtubules are oppositely organized compared to the uniformly oriented microtubules in axons.
Developmental Dynamics: Gradual Enrichment of Minus-End-Out Microtubules in Dendrites
Imaging plus-tip markers provides valuable insights into the dynamic ends of microtubules but offers limited information about the length and stability of the microtubule polymer extending behind the plus end. Therefore, the proportion of comets moving in a particular direction may not directly reflect the precise fraction of microtubule orientations within a given cellular region, as microtubules with different orientations could exhibit variations in length and stability (Fig. 2A). To gain a more detailed understanding of microtubule orientations within a bundled array, we employed live-cell imaging in conjunction with laser-based microsurgery (LS) (Yau et al., 2014). Laser severing of a microtubule bundle generates new microtubule ends, enabling the analysis of newly generated, growing microtubule plus ends (Fig. 2A). By tracking the growth direction of GFP–MT+TIP comets before and after LS, this assay provides a direct measure of microtubule orientations within specific cellular regions, irrespective of microtubule length or plus-end dynamicity (Fig. 2B–D, H–J). An increased proportion of comets moving in a specific direction following LS suggests that microtubules with corresponding orientations are, on average, longer or less dynamic, both indicative of enhanced microtubule stability.
Analyzing the growth direction of GFP–MT+TIP comets before and after LS in axons at different developmental stages (DIV2 to DIV61) consistently revealed that the vast majority of GFP–MT+TIP comets (>95%) exhibited anterograde displacement (Fig. 2G), confirming the uniform plus-end-out microtubule orientation in axons throughout neuronal maturation. We also observed a decrease in the total number of comets in more mature axons, both before and after LS (Fig. 2E,F). This reduction in axonal plus ends may indicate either a decrease in the total number of microtubules within the axon shaft during differentiation, or a replacement of shorter microtubules with longer, more stable microtubules, while maintaining a relatively constant total microtubule number.
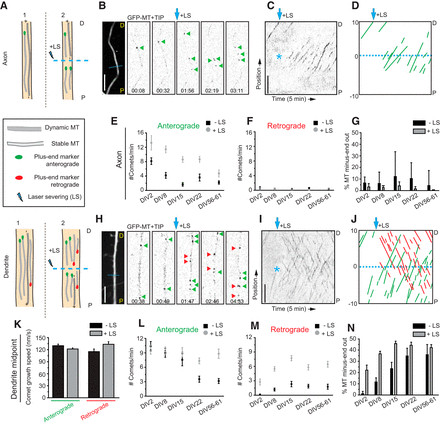 Figure 2.
Figure 2.
In contrast to the uniform axonal microtubule organization, analysis of GFP–MT+TIP comet growth direction and speed in middle segments of primary dendrites revealed the presence of both anterograde and retrograde moving comets across all developmental stages examined (DIV2–DIV61; Fig. 2H–K), consistent with the non-uniform microtubule array in dendrites. The relative proportion of plus-end-in and plus-end-out microtubules in dendrites undergoes significant changes during early dendritic development (DIV2–DIV15) but stabilizes in more mature dendrites (beyond DIV22; Fig. 2N). In young dendrites (DIV2), anterograde displacement of GFP–MT+TIP comets was predominant. However, the appearance of additional retrograde comets following LS (Fig. 2M,N) indicated the presence of stable (long or non-dynamic) minus-end-out microtubules in dendrites even at these early developmental stages, suggesting an early establishment of non-uniformity. In developing dendrites at later stages (DIV8–DIV15), the fraction of plus-end-in comets increased further upon LS, again suggesting that minus-end-out microtubules are, on average, more stable (i.e., longer or non-dynamic; Fig. 2L,M). In mature dendrites (beyond DIV22), the fraction of plus-end-in comets observed before LS increased due to a decrease in the number of plus-end-out comets, indicating that, in mature dendrites, microtubules of both orientations become more stable (i.e., longer or non-dynamic; Fig. 2L,M). These cumulative results robustly demonstrate that mature dendrites are characterized by an approximately equal distribution of plus-end-out and minus-end-out microtubule orientations, highlighting the persistent non-uniformity of dendritic microtubule organization in contrast to the uniform axonal array.
Consistent Mixed Microtubule Orientations Across Dendritic Regions
Previous electron microscopy (EM) studies suggested potential regional differences in microtubule orientation within dendrites, with a reported gradual increase in plus-end-out microtubules in more distal dendritic segments (Baas et al., 1989). As our initial analyses were focused on middle segments of primary dendrites, we expanded our live-cell imaging and LS study to encompass proximal and distal dendritic regions (Fig. 3A) to assess the spatial uniformity or non-uniformity of microtubule organization along the dendrite. In dendrites of DIV15 neurons, we consistently observed mixed microtubule orientations across all dendritic regions examined, with relatively constant ratios of approximately 20% minus-end-out dynamic microtubules (Fig. 3B–D). Following LS, the fraction of minus-end microtubules doubled, indicating that minus-end-out microtubules are more stable (i.e., longer or non-dynamic), while plus-end-out microtubules remain dynamic throughout the entire dendrite. Similarly, LS in mature neurons (DIV55–DIV61) revealed that approximately 40% of microtubules are oriented minus-end-out across proximal, middle and distal dendritic regions (Fig. 3F–I).
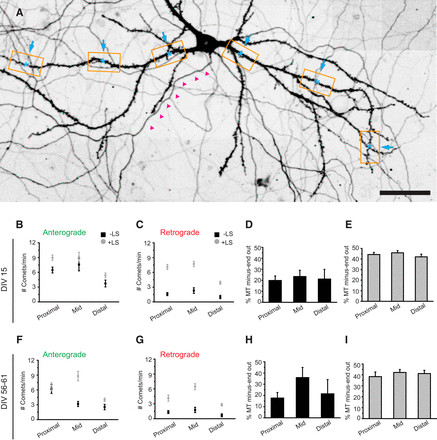 Figure 3.
Figure 3.
We further examined microtubule dynamics in proximal, middle, and distal regions of granule cell dendrites within organotypic hippocampal slice cultures (Fig. 4A–C; Movie 2). Dynamic minus-end-out microtubules were consistently present, albeit in varying numbers, across all three dendritic segments (Fig. 4D; Movie 2). Consistent with our findings in mature dissociated cultures, we observed a slightly elevated ratio of dynamic minus-end-out microtubules in the middle dendritic segment compared to proximal and distal regions (Fig. 4E), suggesting subtle regional variations in microtubule organization within dendrites. To investigate potential position-dependent effects within the middle dendritic segments, defined as 25–60% of total dendritic length, we analyzed microtubule organization in relation to both absolute and relative dendritic length. We found no evidence for differential microtubule organization within these middle segments (Fig. 4F), suggesting a relatively continuous and uniform microtubule organization throughout the middle portions of dendrites, although some variability exists between different dendrites. LS in the same dendritic regions of slice cultures confirmed that the majority of minus-end-out microtubules are also stable in this ex vivo context (Fig. 4H,I).
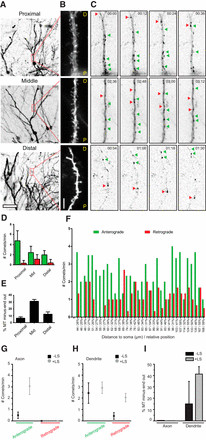 Figure 4.
Figure 4.
These comprehensive analyses across different dendritic regions consistently demonstrate that microtubule orientations in dendrites are mixed throughout their length, further solidifying the notion of non-uniform microtubule organization as a defining feature of dendrites, in contrast to the uniform microtubule array in axons. Our findings also reinforce the earlier observations regarding the differential stability of anterograde and retrograde microtubules in dendrites, with minus-end-out microtubules exhibiting increased stability.
Dendritic Stability: Unveiling Stable Minus-End-Out Microtubules
The increased proportion of plus-end-in comets observed upon LS in dendrites suggested that minus-end-out microtubules are either longer or less dynamic, both indicative of enhanced stability compared to plus-end-out microtubules. To further investigate the stability differences between plus- and minus-end-out microtubules, we meticulously analyzed the precise starting positions of newly formed plus-end microtubules generated after LS. Microtubule severing triggers rapid depolymerization, followed by a rescue event and the formation of a new microtubule plus end, allowing us to assess microtubule orientations and stability. The starting positions of newly formed comets serve as a relative measure of microtubule depolymerization prior to rescue and reflect the resistance of a microtubule to depolymerization, providing insights into microtubule stability in different neuronal compartments. It is important to note that our LS approach does not distinguish between different stable microtubule populations, such as non-dynamic microtubules and long microtubules with dynamic plus ends; both would contribute to increased stability in our assay.
As expected, prior to LS, GFP–MT+TIP comets exhibited a random distribution throughout axons and dendrites in DIV15 cultured hippocampal neurons (Fig. 5A–C). Following microtubule severing in axons, newly formed comets predominantly originated within 0–10 μm proximal to the cut site, with a median starting position at 5.7 μm (Fig. 5D). Similarly, newly formed retrograde comets in dendrites were also concentrated in close proximity to the LS position (median at 5.7 μm; Fig. 5F), indicating comparable stability to axonal microtubules. However, anterograde comets newly formed in dendrites initiated at a significantly greater distance from the cut site (median at 7.9 μm; Fig. 5E), suggesting increased microtubule depolymerization due to the relatively lower stability of anterograde microtubules in dendrites. These findings collectively indicate that minus-end-out microtubules in dendrites exhibit comparable stability to the uniformly stable axonal microtubules, while plus-end-out microtubules in dendrites are comparatively less stable, contributing to the non-uniform stability profile of dendritic microtubules.
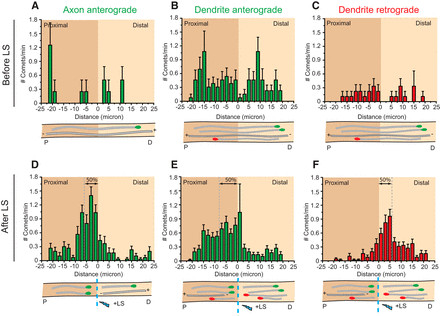 Figure 5.
Figure 5.
Early Neuronal Development: Minus-End-Out Microtubules Present Before Polarization
Previous EM studies proposed that the mixed, non-uniform microtubule organization in dendrites arises after axon initiation and neuronal polarization (Baas et al., 1989, suggesting a developmental switch from uniformity to non-uniformity. However, our observation of minus-end-out microtubules in DIV2 neurons (Fig. 2M,N) prompted us to investigate the precise neurodevelopmental stage at which mixed microtubule polarity is established. We extended our study to stage 2 (non-polarized cells with only neurites) and stage 3 (neurons with an extended axon and minor neurites, which will later differentiate into dendrites; Dotti et al., 1988) neurons. Cortical neurons were nucleofected with GFP–MT+TIP immediately after dissociation, and microtubule growth dynamics were imaged 18–24 h post-transfection, both before and after LS (Fig. 6A–E).
Analysis of GFP–MT+TIP comet growth direction in stage 2 neurites revealed that mixed polarity microtubules are already present even before neuronal polarization. While the majority of comets moved outward, we consistently observed approximately 10% dynamic minus-end-out microtubules in stage 2 neurites, which approximately doubled following LS (Fig. 6G,H). Similar dynamic effects were observed in minor neurites of stage 3 neurons (Fig. 6G,H). However, the approximately 10% minus-end-out microtubules in axons of stage 3 neurons were unaffected by LS, indicating that all axonal minus-end-out microtubules are dynamic at this stage. These results suggest that a stable minus-end-out microtubule population, present in neurites of non-polarized cells, is selectively lost from the axon shortly after neuronal polarization, contributing to the establishment of uniform microtubule polarity in axons and the maintenance of non-uniformity in dendrites.
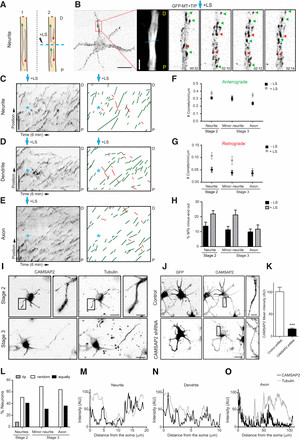 Figure 6.
Figure 6.
Given the role of CAMSAP2 in binding and stabilizing minus ends of non-centrosomal microtubules and its importance for neuronal polarization (Yau et al., 2014), we investigated the localization of CAMSAP2 in neurons before and after axon outgrowth (Fig. 6I). In developing neurons, we observed a distribution of small CAMSAP2 clusters and distinct (punctated) stretches scattered throughout neurites (Fig. 6I–M). Knockdown of CAMSAP2 using established shRNAs (Yau et al., 2014) significantly reduced the antibody signal in stage 2 neurons (Fig. 6J,K), confirming the specificity of CAMSAP2 staining in neurites. Intriguingly, in non-polarized neurons, CAMSAP2 stretches were enriched at some neurite tips (Fig. 6I–M), suggesting a potential role for CAMSAP2 in establishing minus-end-out microtubules in early neurites, contributing to the initial non-uniformity of microtubule organization. At later developmental stages, CAMSAP2 was localized along the axon and minor neurites (Fig. 6I–O). Taken together, these findings demonstrate that mixed microtubule organizations are present in neurites of non-polarized cells, and the formation of uniformly plus-end-out microtubules in axons occurs as a selective process during early axon polarization, contrasting sharply with the persistent non-uniformity of dendritic microtubules.
Taxol-Induced Axonal Processes Exhibit Uniform Plus-End-Out Microtubules
Our data strongly suggest that the selective formation of uniform plus-end-out microtubules is a critical event during axon polarization, contrasting with the inherent non-uniformity of dendritic microtubules. To further examine microtubule dynamics during neuronal polarization, we employed the microtubule-stabilizing drug taxol to induce the formation of multiple axons in cultured neurons (Witte et al., 2008) and analyzed microtubule orientation in these newly formed axonal processes. Treatment with low concentrations of taxol (10 nM) for 3 days significantly increased the number of Tau-positive processes per cell, more than fourfold compared to control neurons (Fig. 7A–D). Neurons were treated with taxol at DIV1, transfected with GFP–MT+TIP at DIV3, and microtubule growth was analyzed before and after LS at DIV4 (Fig. 7D–F). Consistent with our observations in DIV2 and DIV8 neurons (Fig. 2E–G,L,M), GFP–MT+TIP comets in dendrites of non-treated cultures exhibited both anterograde and retrograde displacements, while axons primarily showed anterograde movement (Fig. 7D–I), reflecting the inherent uniformity and non-uniformity of microtubule organization in these compartments, respectively.
However, in axon-like processes induced by taxol treatment, the vast majority of GFP–MT+TIP comets displayed anterograde displacement, with virtually no retrograde comets observed, both before and after LS (Fig. 7G,H). Quantification revealed a near-complete absence of minus-end-out microtubules in taxol-treated neurons, with microtubules overwhelmingly adopting a plus-end-out orientation (Fig. 7I), remarkably similar to the uniform microtubule organization observed in axons of control cells at various developmental stages (Fig. 2E–G). These data demonstrate that taxol treatment nearly eliminates retrograde microtubules in newly induced axonal processes, promoting uniform plus-end-out microtubule polarity. Thus, beyond its known microtubule-stabilizing effects (Witte et al., 2008), the axon-inducing action of taxol appears to be intrinsically linked to the selective formation of uniform plus-end-out microtubules, further emphasizing the critical role of microtubule uniformity versus non-uniformity in neuronal polarization and axonal identity.
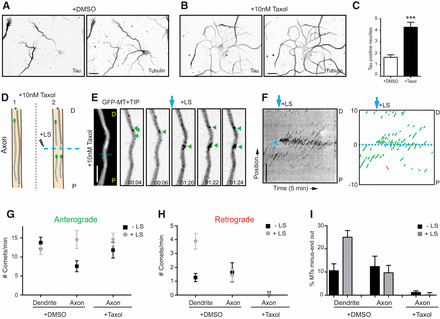 Figure 7.
Figure 7.
Discussion
The distinct microtubule organization in axons and dendrites, characterized by uniformity in axons and non-uniformity in dendrites, has profound implications for neuronal cargo transport and overall neuronal function (Kapitein and Hoogenraad, 2011; Rolls, 2011). While axons typically exhibit microtubules arranged with their plus ends distal to the cell body (uniform plus-end-out), dendrites of mammalian neurons are known to possess a mixed microtubule polarity, containing both plus-end-out and minus-end-out microtubules (Baas and Lin, 2011, highlighting their non-uniformity). Previous studies using second-harmonic generation microscopy in acute hippocampal slices suggested unipolar microtubule arrays in apical dendrites (Dombeck et al., 2003; Kwan et al., 2008), while invertebrate neurons, such as Drosophila, exhibit predominantly minus-end-out microtubule orientation in dendrites (Stone et al., 2008; Goodwin et al., 2012; Maniar et al., 2012). These diverse findings motivated our systematic investigation and comparison of neuronal microtubule dynamics across various mouse and rat model systems, focusing on the contrast between axonal uniformity and dendritic non-uniformity.
Dendritic Microtubule Orientations: Consistent Non-Uniformity Throughout Dendrites
The majority of prior research on neuronal microtubule organization has focused on primary cultured neurons. In this study, we presented a comprehensive analysis of mammalian microtubule organization encompassing in vitro, ex vivo, and in vivo model systems, based on fluorescent microscopic imaging of growing microtubule plus ends. Our findings revealed that dendrites in organotypic slice cultures and in the cortex of living mice exhibit a slight reduction in dynamic microtubule plus ends and growth speed compared to neuron cultures, potentially due to the more complex tissue environments. However, the fundamental ratio of dynamic plus-end-out microtubules (approximately 2:3) and plus-end-in microtubules (approximately 1:3) in dendrites remained remarkably consistent across all model systems, underscoring the conserved nature of dendritic microtubule non-uniformity. Following laser severing of microtubules, we observed an approximately equal ratio (1:1) of plus-end-out and plus-end-in microtubule organization in dendrites, indicating a balanced mixture of both orientations.
Our results align with early EM data from olfactory bulb mitral cells, which demonstrated a consistent mixed microtubule pattern throughout the entire dendrite (Burton, 1988), supporting the notion of dendritic non-uniformity. However, our data diverge from some early EM findings in hippocampal neuron dendrites, which reported uniform plus-end-out microtubule arrays in distal dendritic parts (Baas et al., 1988, 1989). It is important to note that we did observe considerable variation in microtubule orientations across some dendrites (Fig. 4F). Therefore, it is plausible that the limited number of dendrites (n = 3 for position-dependent orientations) analyzed in the original EM studies (Baas et al., 1988, 1989) may have introduced sampling bias, and those reported dendrites may not be representative of the broader neuronal population. Alternatively, given the size and diameter similarity between distal dendrites and axons (Baas et al., 1988, 1989), distinguishing distal dendrites solely based on size measurements in EM samples may be challenging and potentially unreliable.
While mature Drosophila neurons exhibit dendrites with predominantly minus-end-out microtubules, it is noteworthy that developing Drosophila dendrites initially possess mixed microtubule polarity (Hill et al., 2012). Over time, this heterogeneous microtubule array gradually matures into a uniform minus-end-out array. In contrast, our study demonstrates in diverse mammalian neuronal cell types and model systems that microtubules in mature dendrites persistently maintain an antiparallel organization, characterized by non-uniformity. The evolutionary reasons for why mammalian dendrites retain plus-end-out microtubules and do not fully transition to minus-end-out polarity, as observed in Drosophila dendrites, remain an intriguing open question. One possibility is that the presence of oppositely polarized microtubules in dendrites, creating a non-uniform environment, may enhance the efficiency of polarized transport by coordinating the activity of both dynein and specific kinesin family members, allowing for more versatile cargo trafficking within these complex neuronal compartments.
Differential Stability: Distinct Microtubule Stability Profiles in Axons and Dendrites
Our findings demonstrate that axonal microtubules are predominantly stable and oriented plus-end-out across different stages of neuronal development, reinforcing the concept of uniform axonal microtubule organization and stability (Baas et al., 1991; Kollins et al., 2009). In contrast, our data highlight a marked difference in stability between anterograde and retrograde directed microtubules in dendrites, contributing to their overall non-uniform stability profile. Our combined live imaging and laser severing approach allowed us to quantitatively differentiate between stable and dynamic microtubule populations. Based on our data, we propose at least three stages of microtubule organization and stability in dendrites:
- Early Dendritic Growth (DIV2): Growing dendrites in young neurons exhibit a 1:4 ratio of plus-end-in/out microtubule orientations, characterized by stable minus-end-out microtubules and dynamic plus-end-out microtubules, establishing an early form of non-uniformity.
- Developing Dendrites (DIV8–DIV15): While dynamic plus-end-out microtubules become more prominent, dendrites in developing neurons display an almost 1:1 ratio of microtubule orientations, reflecting a more balanced mixture and increasing non-uniformity.
- Mature Dendrites (DIV22–DIV61 and slice cultures): Dendrites in mature neurons contain a slightly higher proportion of stable plus-end-out microtubules, but the overall equal ratio of mixed microtubule orientations and non-uniformity is maintained, indicating a stable heterogeneous microtubule network in mature dendrites.
Microtubule stability in neurons is a complex phenomenon regulated by numerous factors (Kapitein and Hoogenraad, 2015), likely involving the concerted action of diverse microtubule regulatory proteins, including plus-end tracking proteins, minus-end targeting proteins (Akhmanova and Steinmetz, 2008; Akhmanova and Hoogenraad, 2015, microtubule-associated proteins (Dehmelt and Halpain, 2005; Subramanian and Kapoor, 2012, microtubule-severing enzymes (Roll-Mecak and McNally, 2010; Sharp and Ross, 2012, and tubulin-modifying enzymes (Janke and Kneussel, 2010. Future research should focus on elucidating the specific mechanisms responsible for establishing and selectively stabilizing minus-end-out microtubules during dendrite development, contributing to the overall non-uniform microtubule organization in these neuronal compartments.
Microtubule Polarity Establishment: Early Non-Uniformity in Neurites and Axonal Uniformity During Polarization
Early EM studies using hook-decoration techniques suggested that newly formed neurites in non-polarized cells exhibit a uniform plus-end-out microtubule orientation, and the emergence of minus-end-out microtubules in dendritic processes occurs only after axon initiation and neuronal polarization (Baas et al., 1989. These EM data led to a model proposing that the formation of mixed, non-uniform microtubule organization is critical for dendritic differentiation and the establishment of neuronal polarity (Baas and Lin, 2011. However, this model had not been directly tested in living neurons at early developmental stages.
Our findings, in contrast, reveal that mixed polarity microtubules are already present in neurites even before neurons become polarized, demonstrating an early establishment of non-uniformity. We observed that non-polarized neurons contain approximately 20% minus-end-out microtubules in their neurites, indicating an inherent heterogeneity even in nascent neuronal processes. In stage 3 neurons, shortly after axon outgrowth, the microtubule polarity orientation in axons undergoes a significant change: the proportion of minus-end-out microtubules in axons decreases to approximately 10%, while minor neurites (future dendrites) maintain approximately 20% minus-end-out microtubules, suggesting selective elimination of minus-end-out microtubules from the axon during polarization to achieve uniformity. Taxol treatment further corroborated these observations, demonstrating a marked reduction of retrograde microtubules in newly induced axonal processes, promoting uniform plus-end-out polarity. At later developmental stages, the difference in microtubule orientation between axons and dendrites becomes even more pronounced: minus-end-out microtubules almost disappear from axons, while their proportion in dendrites increases to approximately 45%, further emphasizing the divergent pathways towards uniformity in axons and persistent non-uniformity in dendrites.
These distinct microtubule orientations are known to facilitate polarized cargo trafficking in mature neurons (Kapitein and Hoogenraad, 2011; Rolls, 2011). Uniform plus-end-out microtubules in axons enable selective sorting of kinesin-driven vesicles, while minus-end-out microtubules in dendrites allow dynein motors to drive specific cargo to these compartments (Nakata and Hirokawa, 2003; Kapitein et al., 2010b. Interestingly, recent research challenged the contribution of dynein to dendrite-specific transport, suggesting selective (non-axonal) transport already in stage 3, when microtubule orientations were previously thought to be uniformly plus-end-out (Petersen et al., 2014). However, our study provides crucial insights that resolve this apparent contradiction:
- Early Non-Uniformity: Non-polarized stage 2 neurons already contain a significant proportion of minus-end-out microtubules in their neurites, enabling dynein-based transport from the earliest developmental stages and establishing an initial state of non-uniformity.
- Axonal Uniformity Establishment: Axons actively reduce the proportion of stable minus-end-out microtubules in stage 3 neurons by approximately 50%, allowing dynein motors to be excluded from the axon and directing dendritic cargos specifically into future dendrites, actively establishing uniformity in the axon while maintaining non-uniformity in dendrites.
The observation that neurites in non-polarized neurons already contain a mixed population of microtubules, exhibiting non-uniformity, raises fundamental questions about the mechanisms underlying microtubule organization in early neuronal development. How are minus-end-out microtubules generated in these nascent neurites? Potential mechanisms include:
- Centrosomal Nucleation and Transport: Microtubules may be nucleated at the centrosome, subsequently released, and transported into neurites, contributing to both plus-end-out and minus-end-out populations and initial non-uniformity.
- Microtubule Severing: Local katanin or spastin activity may sever microtubules, generating new minus ends and contributing to the mixed polarity and non-uniformity.
- Non-Centrosomal Nucleation: Microtubules may be nucleated at non-centrosomal sites, such as Golgi membranes, local γ-tubulin complexes, or from pre-existing microtubules, potentially generating both plus-end-out and minus-end-out microtubules and contributing to the non-uniformity.
Furthermore, several motor proteins have been implicated in establishing or maintaining minus-end-out microtubule organizations in dendrites. For example, depletion of kinesin-5 increases the proportion of dynamic minus-end-out microtubules, while downregulation of kinesin-6 or kinesin-12 reduces retrograde microtubules in dendrites (Lin et al., 2012; Kahn et al., 2015. Kinesin-1 and kinesin-2 have also been reported to establish uniform minus-end-out microtubules in C. elegans and Drosophila, respectively (Mattie et al., 2010; Yan et al., 2013, suggesting diverse mechanisms for establishing microtubule polarity across species and neuronal compartments, and highlighting the distinct pathways for achieving uniformity or non-uniformity.
Conversely, how are uniform plus-end-out microtubules formed in axons, contrasting with the non-uniformity in dendrites? It is plausible that axons actively eliminate minus-end-out microtubules. Retrograde axonal microtubules may be specifically depolymerized, selectively destabilized, or actively severed. Intriguingly, Tau has been shown to regulate microtubule severing by controlling spastin and katanin activity (Yu et al., 2008; Zempel et al., 2013). Alternatively, axons may selectively stabilize their plus-end-out microtubules, promoting uniformity. Microtubule stabilization in axons is consistent with previous reports of high levels of acetylated tubulin and stable axonal microtubules in stage 3 neurons (Witte et al., 2008). Crucially, local stabilization of microtubules using photoactivatable taxol analogs has been shown to induce axon formation in unpolarized neurons (Witte et al., 2008). Consistently, our findings demonstrate that taxol treatment of cultured neurons generates uniform plus-end-out microtubules by reducing retrograde microtubules in newly formed axonal processes, reinforcing the link between microtubule uniformity and axonal identity.
In conclusion, we propose a model where the selective formation of uniform plus-end-out microtubules in the axon is a critical process underlying neuronal polarization, contrasting sharply with the inherent and persistent non-uniformity of dendritic microtubules. A major challenge for future research is to identify the specific molecular players that interact with the microtubule cytoskeleton and directly control selective microtubule organization, stabilization, and remodeling, ultimately driving axon formation, neuronal polarization, and the establishment of uniformity versus non-uniformity in distinct neuronal compartments.
Footnotes
- This work was supported by the Netherlands Organization for Scientific Research (NWO-ALW-VICI; C.C.H.; NWO-ALW-VIDI; L.C.K.), the Netherlands Organization for Health Research and Development (ZonMW-TOP; C.C.H.), the European Science Foundation (ESF-EURYI; C.C.H.), the European Research Council (ERC-StG; L.C.K.), Swiss National Science Foundation Grant 31003A_135631 (A.H.), the National Centre of Competence in Research (NCCR) SYNAPSY financed by Swiss National Science Foundation Grant 51AU40_125759 (A.H., S.P.), the International Foundation for Research on Paraplegia (A.H.), and the Hans Wilsdorf Foundation (A.H., S.P.). P.S. is supported by an FP7 European Union Marie Curie postdoctoral fellowship and a fellowship of the Swiss National Science Foundation. E.T. received support by the Spanish Education Ministry and a FP7 European Union Marie Curie postdoctoral fellowship. We thank Dr. Akhmanova for helpful discussions and Phebe Wulf for help with cloning constructs. We are grateful to Dr. Didier Trono and Dr. Isabel Bade for providing pSIN-TRE-mSEAP-hPGK-rtTA2sM2 lentivirus vector.
- Correspondence should be addressed to either Casper C. Hoogenraad or Lukas C. Kapitein, Cell Biology, Faculty of Science, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands. c.hoogenraad{at}uu.nl, l.kapitein{at}uu.nl
References
[1](#ref-1) Akhmanova, A., and C. C. Hoogenraad. 2015. Microtubule plus-end tracking proteins: mechanisms and functions. Curr. Biol. 25:R355–R363.
[2](#ref-2) Akhmanova, A., and M. O. Steinmetz. 2008. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 9:309–322.
[3](#ref-3) Baas, P. W., and S. Lin. 2011. Microtubule organization in the axon. J. Neurosci. Res. 89:321–329.
[4](#ref-4) Baas, P. W., J. S. Deitch, M. M. Black, and G. A. Banker. 1987. Polarity orientation of microtubules in hippocampal neurons during axon formation. J. Cell Biol. 105:155–164.
[5](#ref-5) Baas, P. W., M. M. Black, and G. A. Banker. 1988. Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J. Cell Biol. 107:321–330.
[6](#ref-6) Baas, P. W., M. M. Black, K. S.erebro, and G. A. Banker. 1989. Microtubule polarity in the axon and dendrite of hippocampal neurons grown in mass culture. Soc. Neurosci. Abstr. 15:1145.
[7](#ref-7) Baas, P. W., T. P. Pienkowski, and K. S. Kosik. 1991. Processes induced by tau protein and MAP2 are composed of microtubules with stable plus-end-distal polarity orientations. J. Cell Biol. 112:563–572.
[8](#ref-8) Botvinick, E. L., D. J. Cosman, and W. W. Webb. 2004. Fast, scanless laser microsurgery and ablation in live cells with pulsed two-photon excitation. Biophys. J. 87:4176–4188.
[9](#ref-9) Burton, P. R. 1988. Dendrites of mitral cells contain microtubules of opposite polarity. Brain Res. 473:107–115.
[10](#ref-10) Cane, M., F. Kasparavicius, and A. Holtmaat. 2014. In vivo single-cell electroporation for gene delivery to study structural and functional plasticity of cortical circuits. Front. Cell. Neurosci. 8:153.
[11](#ref-11) Colombelli, J., E. Grill, J. Schwab, K. Pepperkok, E. Stelzer, and M. H. Fellner. 2005. Beamsplitter-based laser microdissection. Opt. Express 13:9500–9506.
[12](#ref-12) Conde, C., and A. Cáceres. 2009. Microtubule-based transport, motor proteins, and neuronal polarity. Neuron 63:683–704.
[13](#ref-13) Dehmelt, L., and S. Halpain. 2005. The MAP2/microtubule affinity-regulating kinase (MARK) family of protein kinases. EMBO J. 24:1521–1529.
[14](#ref-14) De Paola, V., L. Holtmaat, O. Knott, S. Song, N. Wilbrecht, R. Caroni, and K. Svoboda. 2003. Cell type-specific structural plasticity of axonal branches and dendritic spines in the neocortex. Neuron 37:611–625.
[15](#ref-15) Dombeck, D. A., K. A. Kasischke, H. D. Vishwasrao, M. Ingelsson, B. T. Hyman, and W. W. Webb. 2003. Second harmonic generation microscopy of ex vivo brain tissue reveals amyloid-beta fibril and congophilic plaque morphology second harmonic generation microscopy of ex vivo brain tissue reveals amyloid-beta fibril and congophilic plaque morphology. J. Biomed. Opt. 8:98–108.
[16](#ref-16) Dotti, C. G., C. A. Sullivan, and G. A. Banker. 1988. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 8:2548–2559.
[17](#ref-17) Goodwin, D., P.ক্তর, P. Hehnly, G. Tanenbaum, and J. R. Fallon. 2012. Complementary roles for Patronin and γ-tubulin in dendrite branching and microtubule organization. J. Neurosci. 32:1308–1319.
[18](#ref-18) Hammond, J. W., S. Slaughter, Y. Li, S. Weissmann, E.R. Verheyden, and P. W. Baas. 2010. Dendrites and axons differ in their intrinsic capacities for microtubule assembly and stabilization. J. Neurosci. 30:3032–3040.
[19](#ref-19) Hill, K. L., A. L. Kenworthy, and L. S. Barlan